GWAS, 즉 Genome-wide association studies는 전장 유전체 분석이라는 분야이다.
최근 GWAS와 연계한 Stroke 연구주제를 시작할 기회가 있어 이제야 듣게 되었는데...
나 또한 굉장히 생소한 개념이라 이에 대해 조금씩 주워들으니 정말 기초부터ㅠ 다시 짚어야되는 부분이 상당히 많았다.
Concept 자체는 모든 유전체 위치에 대해, 내가 연구하고자 하는 형질 및 질병과 연관성을 연구하는 것인데 아직까진 감이 잘 안 온다.
이에 대해 두마디 정밀 의료라는 블로그에서 정말 자세히 설명을 많이 해주셨는데,
GWAS는 일반적으로 Case (관심 형질을 가진 집단; 환자군) 와 Control (형질을 갖지 않는 집단; 정상군)의 두 집단의 유전 정보를 얻은 후에 서로 비교하여, case에서 더 많은 빈도를 갖는, 즉 연관성을 가진 유전자를 찾게 됩니다. 한 가지 중요한 내용은 GWAS에서 찾아낸 유전자라 하더라도, 그것이 항상 원인 유전자는 아니라는 점입니다. 즉 GWAS는 인과 관계를 찾는 것이 아니라 우연히 연관되어 나타나는 유전자들의 후보를 찾는 과정입니다.
라는 말씀을 해주셨다. 우연한 기회로 시간이 허락해 좀 더 GWAS에 대해 자세히 알아보는 시간을 Natrue review article을 보며 짚어보고자 한다. 참고로 GWAS에 대한 Data는 인터넷에 오픈소스가 굉장히 많은데, 보통은 summarized data로 특정 phenotype과 관계된 SNP들의 P value와 beta 값 (effect size)가 공개되어 있다. (https://www.ebi.ac.uk/gwas/ GWAS CATALOG 참조)
(만약 연구자가 raw data를 가지고 이러한 GWAS 연구를 하고자 한다면 어떠한 SNP들이 특정 형질과 관계가 있는지 추정하는 통계적 방법들도 여럿이 있다. Rough하게 살펴보자면 single-variant analysis, Polygenic Risk Score, Machine learning approaches - LASSO, RIDGE... , Bayesian mehods... 들이 있다. CT clumping을 통해 LD가 높은 SNP들을 정리하고 독립적인 유전적 신호만을 선택해 PRS의 정확도를 높이기도 한다.)
GWAS에 대해 이해하는데 필요한 핵심 개념들이 여러개가 있는데, 그 중 SNP(Single Nucleotide Polymorphism) 와 LD(Linkage Disequilibrium) Block에 대해 우선 알아보자.
SNP는 DNA 염기서열 중 하나의 서열에 돌연변이가 생기며 이후 집단 내 일정 빈도로 존재하는 유전변이인데, GWAS는 보통 10만개에서 100만 개 이상의 SNP들을 Microarray를 통해 (정말 mechanical하게) 개개인의 SNP Genotype들을 결정하고, 이 중 질병이나 phenotype과 가장 관련성을 높게 나타내는 SNP를 찾아내는 분석이다.
실제 GWAS를 해보면 P value가 일반 의학 논문들에서 보던것과는 상이한 것을 알 수 있는데, 이는 통계적으로 중복비교의 문제가 생기기 때문이다. 50만개의 SNP를 하나의 표현형에 대해 분석을 한다면, 중복비교에 대한 보정을 한 이후 1 x 10^-7 이하의 유의수준을 보이는 경우 유의한 유전변이로 판정할 수 있다. 이렇게 밝혀진 SNP들은 transferability 와 같이 다른 인종에서도 유의성 여부를 검정하기도 한 후 GWAS catalogue에서 확인할 수 있다.
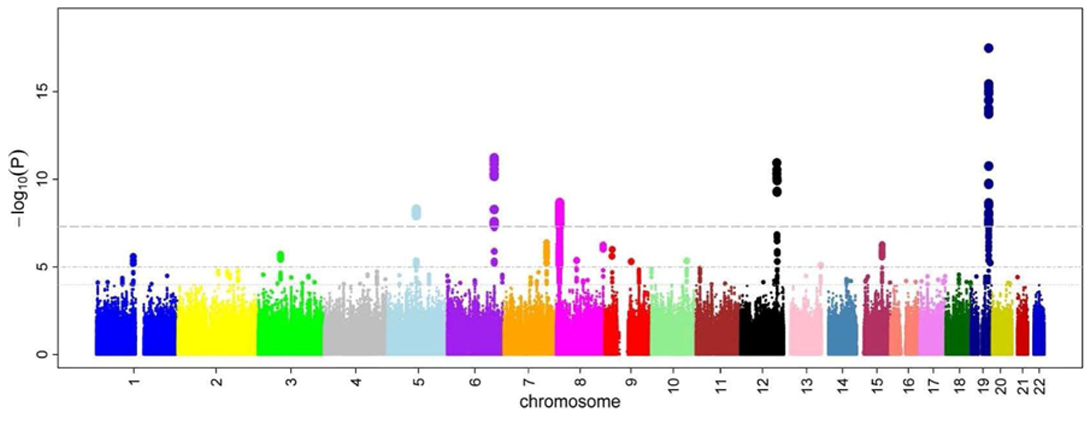
앞서 발견된 SNP들은 phenotype에 영향을 미치는 원인유전변이일수도 있지만 다른 유전변이들에 대한 marker로서의 역할도 할 수 있다. 후자의 경우에는 실제적 원인유전변이와 높은 LD 상태에 있는 것으로 받아들여지는데, Meiosis, 즉 감수분열 때 두 개의 SNP들이 Crossover가 일어나 각각 따로 유전될 수도 있음에도 불구하고 해당 SNP들이 염색체 상에서 서로 가까이 위치하기 때문에 함께 유전될 확률이 높아지는 것이다.
LD의 정도는 LD score regression으로 분석하게 되는데, 각 SNP의 주변 SNP들과의 LD 값을 R^2으로 합산해서 LD score로 나타낸다. 같은 LD Block에 포함된 위치에 대해서는 동일한 연관성을 시사하는 p값을 보이기 때문에, 해당 block의 대표적인 하나의 마커만 사용해도 되니 분석 위치의 수를 줄일 수 있다. 마찬가지로 후보 loci를 찾았다 하더라도 이는 LD block 내 존재한 다른 위치일 수 있다. 그리고 위의 manhattan plot에서 signal이 하나만 높게 솟은 게 아니고 주변에도 높은 signal을 보이는 이유가 된다. Imputation이라고 부르는 과정을 통해 같은 LD Blcok 내의 검사하지 않은 부위의 유전형도 추정이 가능해지는데, 연구 대상자의 일부 유전형이 직접적으로 측정된 데이터가 있다면 이를 Referecne panel의 LD Block을 이용하여 원래 존재하지 않았던 SNP data를 생성할 수 있게 된다. 이를 통해 GWAS의 해상도를 높일 수 있다.
새롭게 SNP를 찾았다면 크게 네 가지 방법으로 이를 분석할 수 있는데, 첫 번째로는 발굴된 SNP와 기존 보고된 GWAS 결과들의 관련성을 보는 방법이다. 두 SNP들이 높은 LD를 보이는 경우에는 발굴된 SNP와 LD상에 있는 보고된 SNP는 같은 유전적인 영향력을 가지는 것으로 생각할 수 있다. 두 번째로는 SNP가 위치하는 부위에서 근접하게 존재하는 유전자들에서 대해 특정 phenotype과 관련성을 찾는 방법이다. 세 번째로는 ENCODE (Encyclopedia of DNA elements) 프로젝트의 결과를 활용하는 해석이다. 전사인자결합부위, 크로마틴 구조변화, 히스톤 단백질 메틸화 혹은 아세틸화 등의 정보가 존재한다면, 해당 SNP는 유전자 발현 조절에서 역할을 추정한다. 특정 유전자의 발현이 SNP의 유전자형에 따라 변화된다면, 그 SNP를 유전자의 eQTL(expression quantitative trait loci)이라 부른다. 마지막으로는 발견된 SNP들의 인종 간 대립인자 빈도를 비교함으로써 해당 SNP의 인종 특이적 관련성 여부 및 진화적 중요성을 이해할 수 있다.
왜 GWAS Catalogue에 있는 SNP들만 찾아서 연구를 하면 되지 않느냐고 반문할 수 있지만, 이는 발굴된 유전변이들의 약 90%는 유전자의 인트론 영역이나 애초에 유전자가 없는 영역에 있는 변이들로 단순히 연관성만이 높기 때문에 찾아진 경우가 대부분이기 때문이다. 이런 것들을 극복하기 위해 NGS (Next Generation Sequencing)을 황용해 집단 내 저빈도 (1%)로 존재하는 유전변이들까지 포함한 모든 유전변이를 발굴하고 표현형과의 관련성을 보기도 한다.
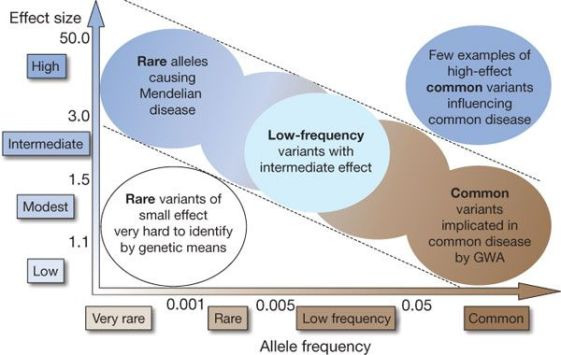
GWAS는 분명 유용한 수단이지만 단지 상관관계만을 알 수 있지 이것이 인과관계는 설명하지 못한다는 약점이 있다. 임상연구에 종사하는 전문가들의 연구 주제 선정이 중요한 이유 중 하나다.
다음 글에서는 Nature에서 2021년 publish 된 GWAS review article에 대해 살펴보며 GWAS의 전반적 과정에 대해 살펴보고 Stroke에서의 활용은 어찌되고 있는지 간략한 history를 다뤄보려 한다.
** 헷갈리는 개념 정리
Effect size (베타값)은 특정 SNP가 형질에 미치는 영향을 나타내고, Heritability (유전률)은 특정 형질의 변이가 유전적 요인에 의해 얼마나 설명되는지를 나타낸다. 전체 변이 중 요전적 요인이 차지하는 비율을 의미
Ref )
Uffelmann, E., Huang, Q.Q., Munung, N.S. et al. Genome-wide association studies. Nat Rev Methods Primers 1, 59 (2021). https://doi.org/10.1038/s43586-021-00056-9
Mishra, A., Malik, R., Hachiya, T. et al. Stroke genetics informs drug discovery and risk prediction across ancestries. Nature 611, 115–123 (2022). https://doi.org/10.1038/s41586-022-05165-3
Malik, R., Chauhan, G., Traylor, M. et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet 50, 524–537 (2018). https://doi.org/10.1038/s41588-018-0058-3
Lee KJ, Kim H, Lee SJ, Duperron MG, Debette S, Bae HJ, Sung J. Causal Effect of the 25-Hydroxyvitamin D Concentration on Cerebral Small Vessel Disease: A Mendelian Randomization Study. Stroke. 2023 Sep;54(9):2338-2346. doi: 10.1161/STROKEAHA.123.042980. Epub 2023 Jul 19. PMID: 37465996; PMCID: PMC10453327.
전장 유전체 연관 분석, GWAS란 무엇인가?
어제 정신과 전문의 친구와 점심을 먹었습니다. 제가 병원 연구실에서 유전체 연구를 하는 것을 듣고, 함께 연구할 아이디어에 대해서 이야기를 나누자고 만났는데, 안타깝게도 GWAS에 대한 개념
2wordspm.wordpress.com
홍경원, The Introduction and Prospect of Genome-Wide Association Study, Public Health Weekly Report, KCDC 제 7권 제 33호
https://youtu.be/Pdic7p_dk0I?si=CoLS4uVkZY9Fxs6K
'GENETICS' 카테고리의 다른 글
| Mendelian Randomization - Essential for study design (2) | 2024.09.20 |
|---|---|
| LD Block 에 대해 (0) | 2024.09.12 |
| PRS calculation (2024 GENESIS-K 유전체 강의 정리) (4) | 2024.08.26 |
| GWAS의 Application (3) (0) | 2024.08.24 |
| GWAS 의 전반적 과정 (2) (0) | 2024.08.22 |




댓글